SEQMOL index
Measuring physical residue covariation between two columns of sequence alignment
Download
When two residues in a sequence alignment are suspect for forming a physical interaction, and the protein structure is not available, one could use physical covariation module to evaluate this possibility.
Physical covariation module requires, at the minimum, only sequence alignment. PDB file is not necessary.
With co-variation module turned on ![]() , two residues are chosen in a sequence alignment by double-clicking. The covariation and quality of the residue pairs will be determined for all the aligned sequences and the resulting pairing score will be produced. It is possible to fix on one residue and then evaluate a range of other residues against this one residue automatically:
, two residues are chosen in a sequence alignment by double-clicking. The covariation and quality of the residue pairs will be determined for all the aligned sequences and the resulting pairing score will be produced. It is possible to fix on one residue and then evaluate a range of other residues against this one residue automatically: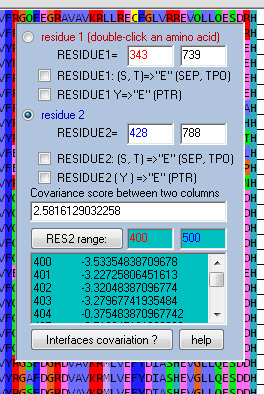
Example.
Covariation scan of sequence alignment for residue 717 pairing with residue range: 680-750: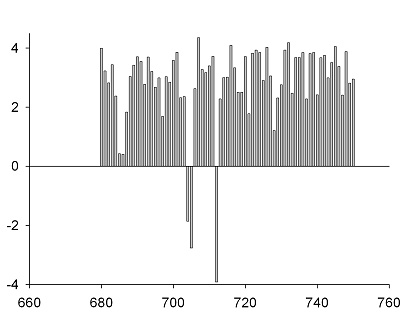
Interpretation.
Unlikely 3D partners of residue 717:
(scores near 0 or negative)
No. Score
685 0.424032
686 0.412258
704 -1.85823
705 -2.76597
712 -3.91806
No. Score
707 4.351452
732 4.181935
716 4.092258
745 4.054032
726 4.026129
These scores were generated from multiple sequence alignment alone. For this particular example, crystal structure of the protein sequence No. 42 is available and it is possible to verify the analysis. True 3D partners of residue 717 (green color shows correct predictions):
707 4.1A
708 3.7A
711 4.2A
716 4.3A
719 3.2A
(for positions 708, 711 and 719 the scores were still very positive but far enough from the top to not result in useful predictions: 3.275645, 3.724355, 2.50629)
Co-variation analysis thus found 2 out of 5 proximal residues of position 717 placing them in top 4% of all 70 residues scanned, and correctly ruled out several positions that are not spatially close to 717. All without knowing anything about the PDB structure.
Several residue interaction matrices can be used in the analysis.
When 3D structure(s) of proteins are known, this analysis can be extended to evaluate interactions of poorly conserved protein regions or protein-protein complexes.